
© Janssen Biotech, Inc. 2021. All rights reserved.
cp-43578v4
No footer content fetched
For US Healthcare Providers
For US Healthcare Providers
Choose A Condition:
PLEASE SELECT
ACR20 Response at Week 14 (primary endpoint): 75% of patients receiving SIMPONI ARIA® +/- MTX achieved ACR20 response vs 22% of patients receiving placebo +/- MTX (P<0.001)1-3
Greater reduction in enthesitis score vs placebo:
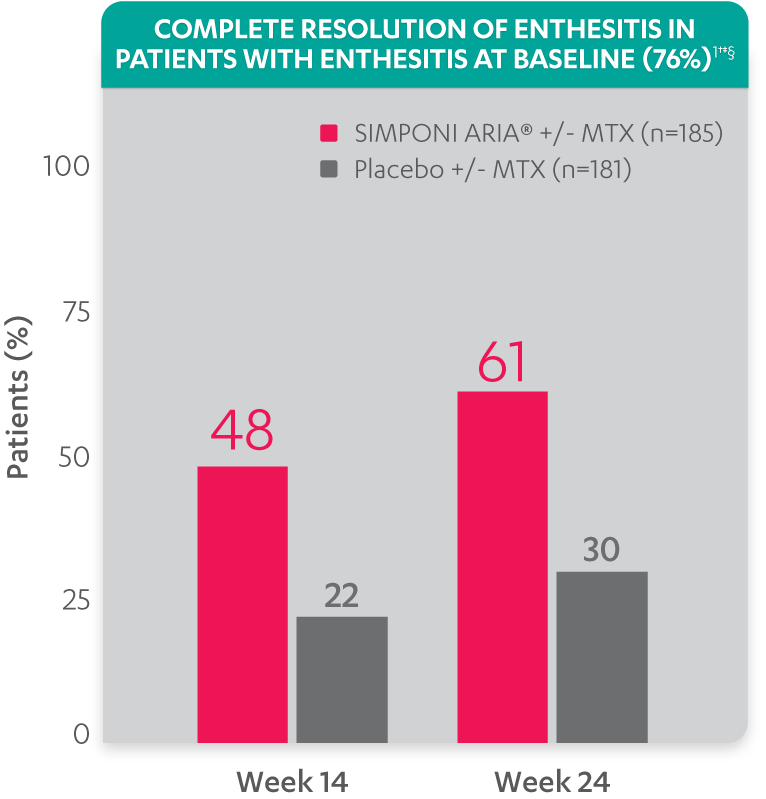
Resolution of enthesitis at Week 14 and Week 24 was not adjusted for multiplicity. Therefore, statistical significance has not been established.
*No missing data imputation rule is applied.
†A decrease in Leeds Enthesitis Index (LEI) score indicated improvement. Only patients with enthesitis at baseline were included in the LEI analyses. The LEI index ranges from 0 to 6. Patients with enthesitis at baseline had an LEI enthesitis score >0 at Week 0. Patients with resolution of enthesitis had a score of 0 at the time point being analyzed. 76% of patients had enthesitis at baseline.
‡Resolution of enthesitis is based on imputed data using LOCF for missing data.
§The same patients may not have responded at each time point.
Study design: GO-VIBRANT was a global, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of SIMPONI ARIA® compared with placebo in 480 adult patients with active PsA. The target study population was biologic-naïve patients with active PsA for ≥6 months who met ClASsification criteria for Psoriatic ARthritis (CASPAR) criteria at screening. Patients in this trial had a diagnosis of PsA for at least 6 months and had symptoms of active disease (≥5 swollen joints and ≥5 tender joints and a CRP level of ≥0.6 mg/dL). At Week 0, patients were randomized in a 1:1 ratio to 1 of 2 treatment groups. Patients were allowed to be treated with or without MTX. Patients in the placebo group (n=239) were randomized to receive IV placebo infusions at Weeks 0, 4, 12, and 20. Patients in the SIMPONI ARIA® group (n=241) were randomized to receive SIMPONI ARIA®2 mg/kg infusions at Weeks 0, 4, and q8w thereafter through Week 52. Patients were to receive a placebo infusion at Week 24 to maintain the treatment blind. At Week 24, all patients switched to treatment with SIMPONI ARIA®2 mg/kg and were to receive administrations at Weeks 24, 28, and q8w through Week 52. The primary endpoint was the percentage of patients achieving an ACR20 response at Week 14.
ACR20 Response at Week 14 (primary endpoint): 75% of patients receiving SIMPONI ARIA® +/- MTX achieved ACR20 response vs 22% of patients receiving placebo +/- MTX (P<0.001)1-3
Greater reduction in dactylitis score vs placebo:
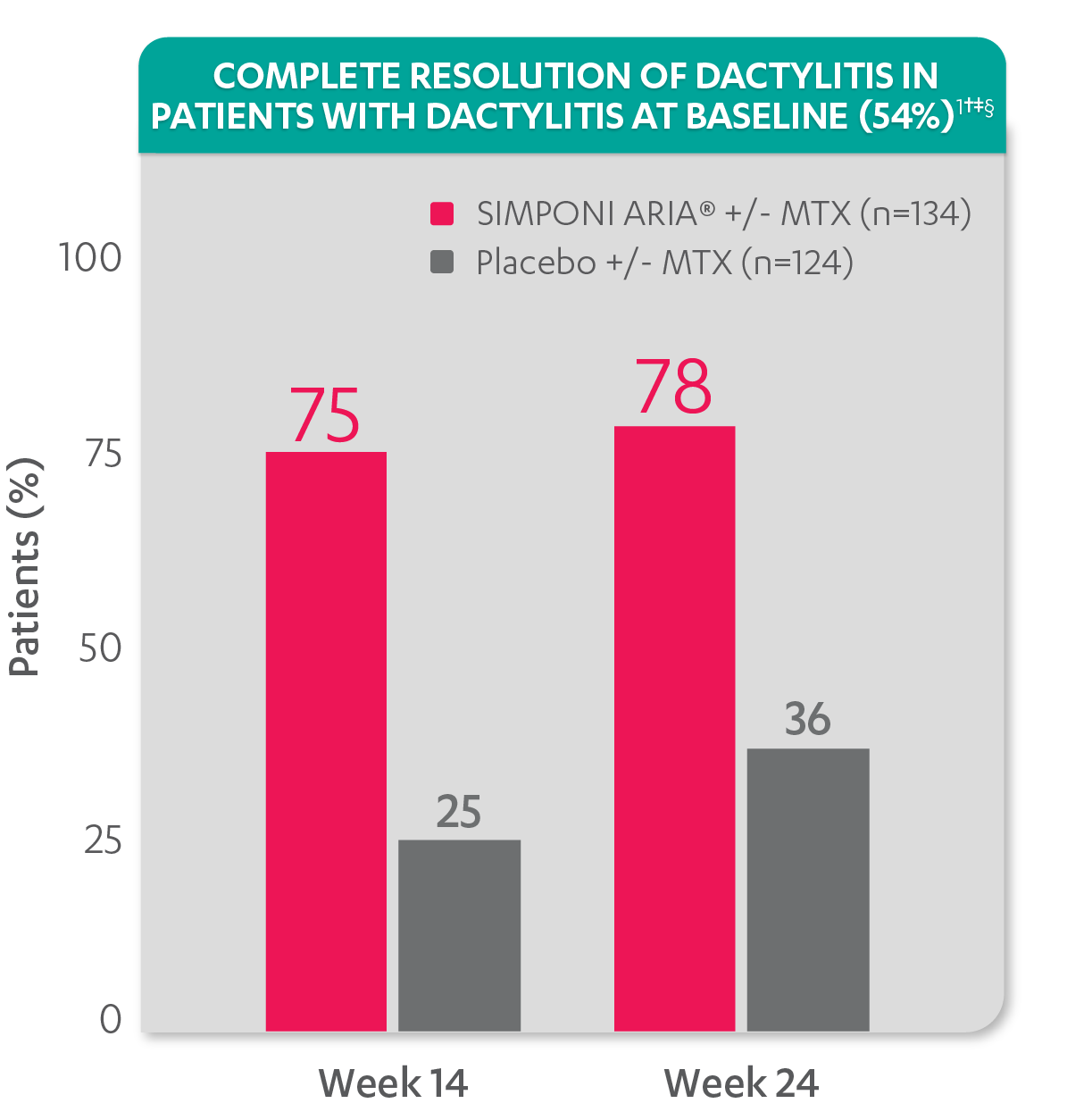
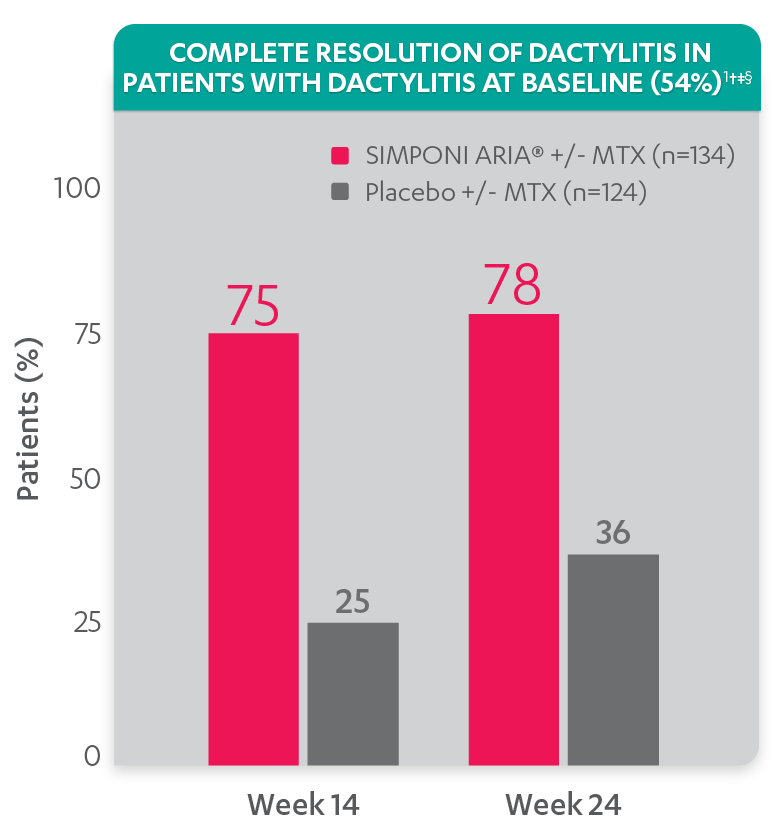
Resolution of dactylitis at Week 14 and Week 24 was not adjusted for multiplicity. Therefore, statistical significance has not been established.
*No missing data imputation rule is applied.
†A decrease in dactylitis score indicated improvement. Only patients with dactylitis at baseline were included in the dactylitis analyses. The dactylitis index ranges from 0 to 60. Patients with dactylitis at baseline had a dactylitis score >0 at Week 0. Patients with resolution of dactylitis had a score of 0 at the time point being analyzed. 54% of patients had dactylitis at baseline.
‡Resolution of dactylitis is based on imputed data using LOCF for missing data.
§The same patients may not have responded at each time point.
Study design: GO-VIBRANT was a global, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of SIMPONI ARIA® compared with placebo in 480 adult patients with active PsA. The target study population was biologic-naïve patients with active PsA for ≥6 months who met ClASsification criteria for Psoriatic ARthritis (CASPAR) criteria at screening. Patients in this trial had a diagnosis of PsA for at least 6 months and had symptoms of active disease (≥5 swollen joints and ≥5 tender joints and a CRP level of ≥0.6 mg/dL). At Week 0, patients were randomized in a 1:1 ratio to 1 of 2 treatment groups. Patients were allowed to be treated with or without MTX. Patients in the placebo group (n=239) were randomized to receive IV placebo infusions at Weeks 0, 4, 12, and 20. Patients in the SIMPONI ARIA® group (n=241) were randomized to receive SIMPONI ARIA® 2 mg/kg infusions at Weeks 0, 4, and q8w thereafter through Week 52. Patients were to receive a placebo infusion at Week 24 to maintain the treatment blind. At Week 24, all patients switched to treatment with SIMPONI ARIA® 2 mg/kg and were to receive administrations at Weeks 24, 28, and q8w through Week 52. The primary endpoint was the percentage of patients achieving an ACR20 response at Week 14.