Choose A Condition:
Please Select
FOR ADULTS WITH ACTIVE PsA
ACR20 RESPONSE RATES OVER TIME
75%

22%

ACR20 response in patients receiving SIMPONI ARIA® + MTX was
similar to those receiving SIMPONI ARIA® without MTX1-4
46%
8%
77%
24%
ACR20 responses at Week 2 were not adjusted for multiplicity. Therefore, statistical significance has not been established.

77%
77%
*The same patients may not have responded at each time point.
†ACR20 responses are based on imputed data using treatment failure, last observation carried forward for partially missing data, and nonresponder imputation for completely missing data.
‡In this ITT analysis, patients were considered to be nonresponders if they experienced any of the following: increased the dose of MTX or oral corticosteroids over baseline for PsA not as part of early escape; initiated treatment with DMARDs, systemic immunosuppressives, treatment with oral, IV, or intramuscular corticosteroids, and/or biologics for PsA; met early escape criteria at Week 16 and started concomitant medication intervention; or discontinued treatment due to an unsatisfactory therapeutic effect.
§Treatment failure criteria were not applied after Week 24.
||At Week 24, all remaining patients in the placebo +/- MTX group began receiving SIMPONI ARIA® +/- MTX in a blinded manner. At Week 24, subjects in the SIMPONI ARIA® +/- MTX group received a placebo infusion to maintain the treatment blind, and then continued to receive SIMPONI ARIA® +/- MTX at Week 28 and q8w thereafter through Week 52.
¶After Week 24, selected sponsor personnel were unblinded to subject-level data, which may have affected results.
ACR50 RESPONSE RATES OVER TIME
Major Secondary Endpoint
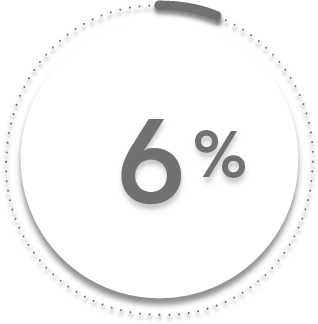
44%

6%
13%
0.4%
54%
6%
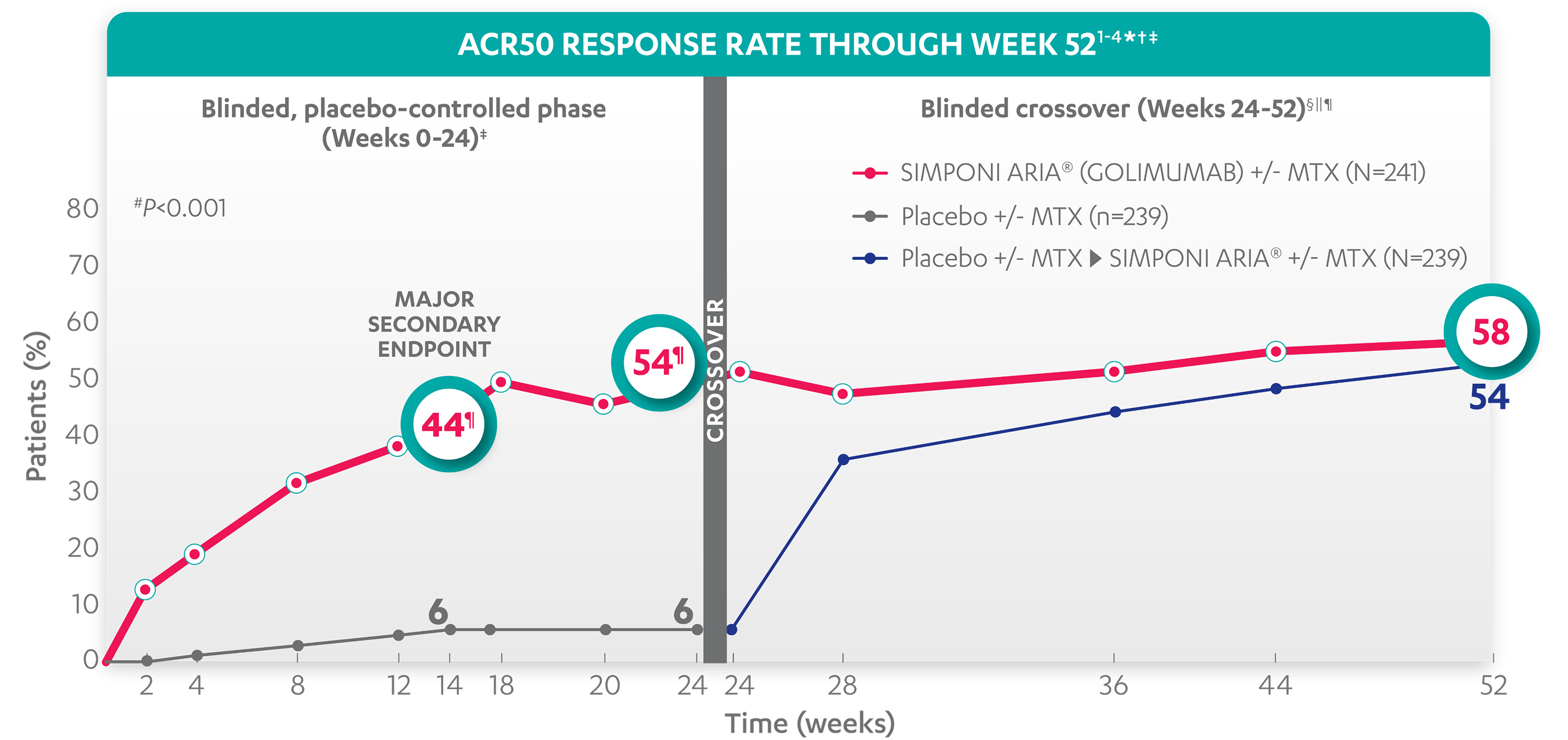
58%
54%
*The same patients may not have responded at each time point.
†ACR50 response is based on imputed data using treatment failure, last observation carried forward (LOCF) for partially missing data, and nonresponder imputation (NRI) for completely missing data.
‡In this intention-to-treat (ITT) analysis, patients were considered to be nonresponders if they experienced any of the following: increased the dose of MTX or oral corticosteroids over baseline for PsA not as part of early escape; initiated treatment with DMARDs, systemic immunosuppressives, treatment with oral, intravenous (IV), or intramuscular (IM) corticosteroids, and/or biologics for PsA; met early escape criteria at Week 16 and started concomitant medication intervention; or discontinued treatment due to an unsatisfactory therapeutic effect.
§Treatment failure criteria were not applied after Week 24.
||At Week 24, all remaining patients in the placebo +/- MTX group began receiving SIMPONI ARIA® +/- MTX in a blinded manner. At Week 24, subjects in the SIMPONI ARIA® +/- MTX group received a placebo infusion to maintain the treatment blind, and then continued to receive SIMPONI ARIA® +/- MTX at Week 28 and q8w thereafter through Week 52.
¶After Week 24, selected sponsor personnel were unblinded to subject-level data, which may have affected results.
ACR70 RESPONSE RATES OVER TIME
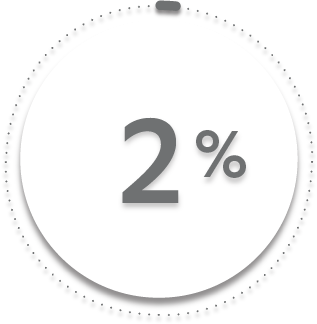
25%

2%
33%
3%
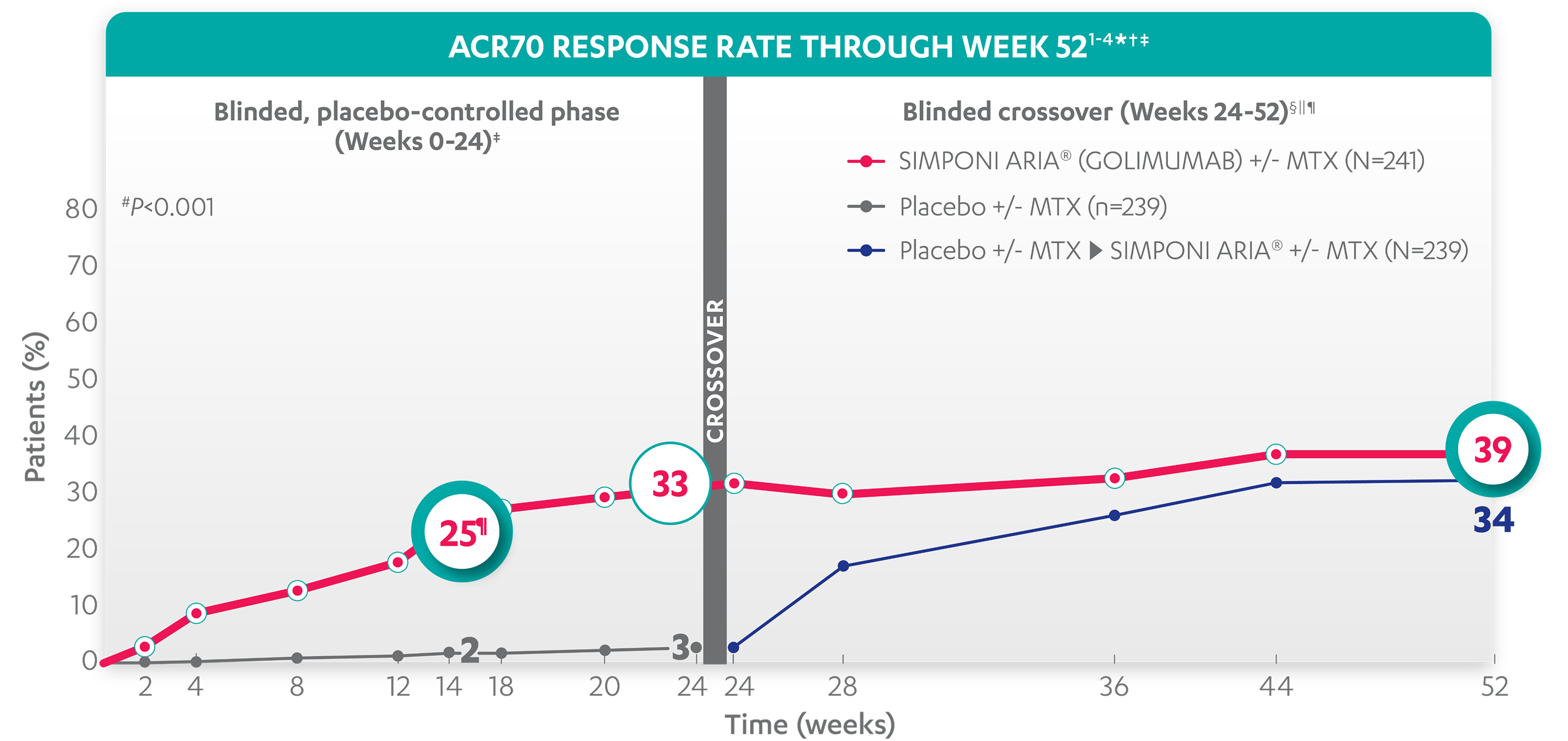
39%
34%
*The same patients may not have responded at each time point.
†ACR70 response is based on imputed data using treatment failure, last observation carried forward (LOCF) for partially missing data, and nonresponder imputation (NRI) for completely missing data.
‡In this intention-to-treat (ITT) analysis, patients were considered to be nonresponders if they experienced any of the following: increased the dose of MTX or oral corticosteroids over baseline for PsA not as part of early escape; initiated treatment with DMARDs, systemic immunosuppressives, treatment with oral, intravenous (IV), or intramuscular (IM) corticosteroids, and/or biologics for PsA; met early escape criteria at Week 16 and started concomitant medication intervention; or discontinued treatment due to an unsatisfactory therapeutic effect.
§Treatment failure criteria were not applied after Week 24.
||At Week 24, all remaining patients in the placebo +/- MTX group began receiving SIMPONI ARIA® +/- MTX in a blinded manner. At Week 24, subjects in the SIMPONI ARIA® +/- MTX group received a placebo infusion to maintain the treatment blind, and then continued to receive SIMPONI ARIA® +/- MTX at Week 28 and q8w thereafter through Week 52.
¶After Week 24, selected sponsor personnel were unblinded to subject-level data, which may have affected results.
References: 1. Data on file. Johnson & Johnson. 2. SIMPONI ARIA® (golimumab) [Prescribing Information]. Horsham, PA: Johnson & Johnson. 3. Kavanaugh A, Husni ME, Harrison DD, et al. Safety and efficacy of intravenous golimumab in patients with active psoriatic arthritis: results through week twenty-four of the GO-VIBRANT study. Arthritis Rheumatol.
2017;69(11):2151-2161. doi: 10.1002/