
© Janssen Biotech, Inc. 2021. All rights reserved.
cp-43578v4
No footer content fetched
For US Healthcare Providers
For US Healthcare Providers
Choose A Condition:
PLEASE SELECT
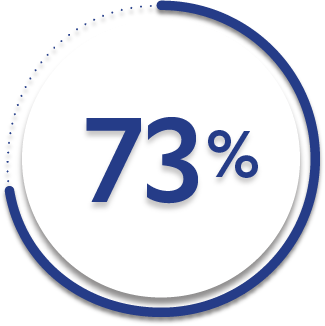
73%

VS
26%

37%
VS
19%
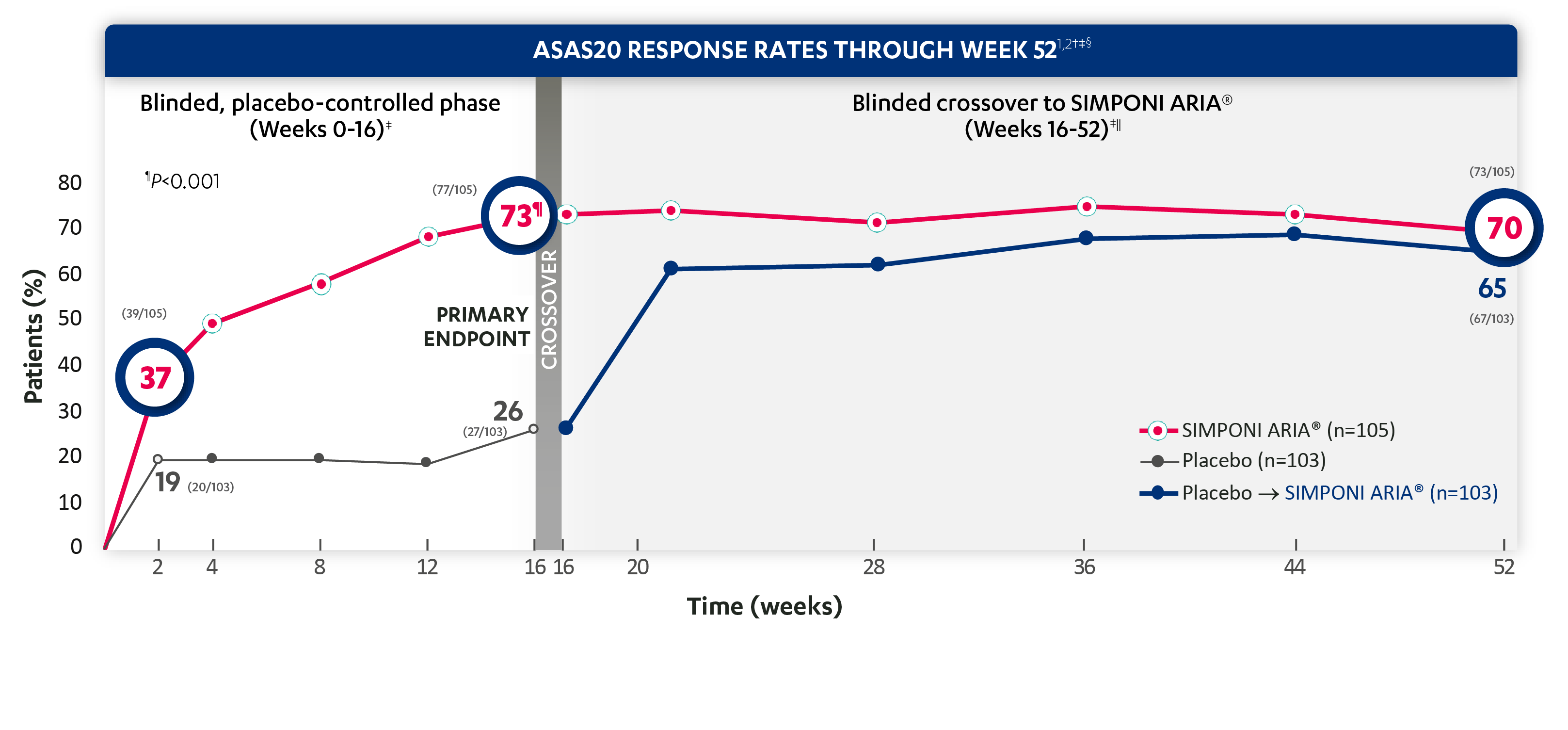
ASAS20 response at Week 2 was not adjusted for multiplicity. Therefore, statistical significance was not established.
*ASAS=Assessment of SpondyloArthritis international Society. An ASAS20 response is defined as an improvement of ≥20% from baseline and absolute improvement from baseline of at least 1 on a 0-to-10 scale in at least 3 of the following 4 domains: patient global, total back pain, function (BASFI), and inflammation (average of the last 2 questions of the BASDAI concerning morning stiffness). An absence of deterioration from baseline (deterioration defines as ≥20% worsening and absolute worsening of at least 1 on a 0-to-10 scale) in the potential remaining domain. An ASAS40 response is defined as a ≥40% improvement in 3 of the 4 domains with an absolute improvement of at least 2 on a 0-to-10 scale, and no worsening in the remaining domain.
71%
VS
62%
70%
VS
65%
ASAS20 response at Week 2 was not adjusted for multiplicity. Therefore, statistical significance was not established.
†The same patients may not have responded at each time point.
‡ASAS20 response is based on imputed data using treatment failure (only through Week 16), LOCF for partially missing data, and NRI for completely missing data.
§In this intention-to-treat (ITT) analysis, patients were considered to be nonresponders if they experienced any of the following: initiated new DMARDs, biologics, or systemic immunosuppressives; increased SSZ, MTX, or HCQ above baseline dose; initiated treatment with oral, IV, or IM corticosteroids; increased the dose of oral corticosteroids above baseline dose; or discontinued treatment due to unsatisfactory therapeutic effect. Treatment failure rules were applied only through Week 16. Last observation carried forward (LOCF) rules were applied for partially missing data.
||At Week 16, all remaining patients in the placebo group began receiving SIMPONI ARIA® in a blinded manner.
Study design: GO-ALIVE was a global, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of SIMPONI ARIA® compared with placebo in 208 adult patients with active AS with an inadequate response or intolerance to NSAIDs. Patients had a diagnosis of definite AS for at least 3 months according to modified New York criteria. Patients had symptoms of active disease [Bath AS Disease Activity Index (BASDAI) ≥4, VAS for total back pain of ≥4, on scales of 0 to 10 cm (0 to 100 mm), and a hsCRP level of ≥0.3 mg/dL (3 mg/L)]. At Week 0, patients were randomized in a 1:1 ratio to 1 of 2 treatment groups. Subjects in the placebo group (n=103) were randomized to receive IV placebo infusions at Weeks 0, 4, and 12. At Week 16, these patients were crossed over to SIMPONI ARIA® and received administrations at Weeks 16, 20, and q8w thereafter through Week 52. Patients in the SIMPONI ARIA® group (n=105) were randomized to receive SIMPONI ARIA® 2 mg/kg infusions at Weeks 0, 4, and 12. These patients received a placebo infusion at Week 16 to maintain the treatment blind and continued to receive SIMPONI ARIA® infusions at Week 20 and q8w thereafter through Week 52. Patients were allowed to continue stable doses of concomitant MTX, SSZ, hydroxychloroquine (HCQ), low dose oral corticosteroids (equivalent 25 to ≤10 mg of prednisone per day), and/or NSAIDs during the trial. The primary endpoint was the percentage of patients achieving ASAS20 response at Week 16.
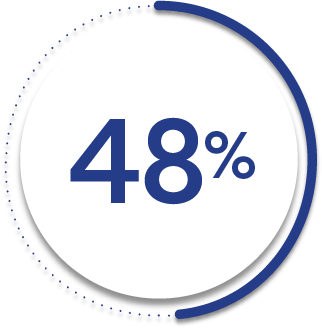
48%
VS
9%

14%
VS
4%
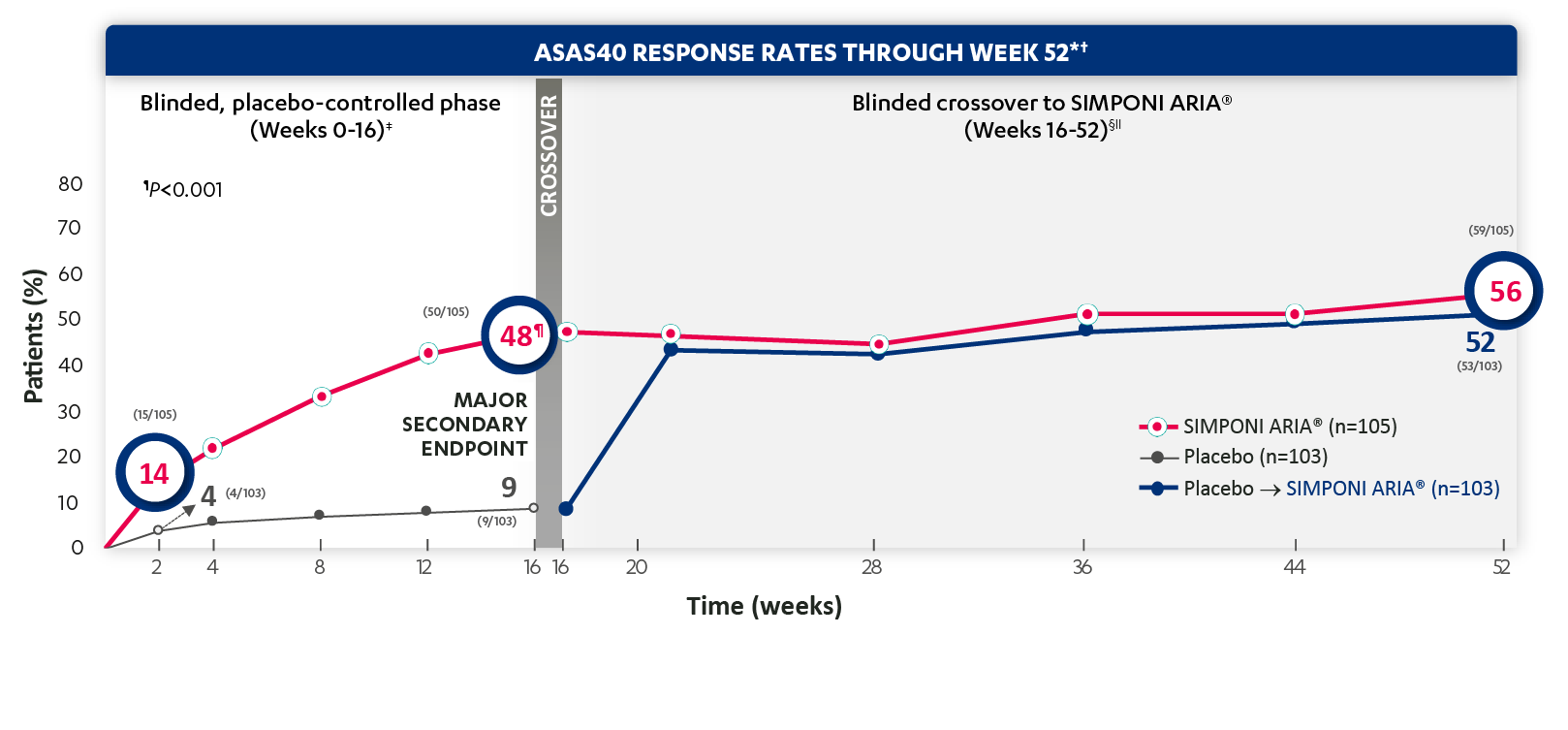
ASAS40 response at Week 2 was not adjusted for multiplicity. Therefore, statistical significance was not established.
VS
VS
ASAS40 response at Week 2 was not adjusted for multiplicity. Therefore, statistical significance was not established.
*ASAS=Assessment of SpondyloArthritis international Society. An ASAS20 response is defined as an improvement of ≥20% from baseline and absolute improvement from baseline of at least 1 on a 0-to-10 scale in at least 3 of the following 4 domains: patient global, total back pain, function (BASFI), and inflammation (average of the last 2 questions of the BASDAI concerning morning stiffness). An absence of deterioration from baseline (deterioration defines as ≥20% worsening and absolute worsening of at least 1 on a 0-to-10 scale) in the potential remaining domain. An ASAS40 response is defined as a ≥40% improvement in 3 of the 4 domains with an absolute improvement of at least 2 on a 0-to-10 scale, and no worsening in the remaining domain.
†ASAS40 response is based on imputed data using treatment failure (only through Week 16), Last observation carried forward (LOCF) for partially missing data, and NRI for completely missing data.
‡The same patients may not have responded at each time point.
§In this intention-to-treat (ITT) analysis, patients were considered to be nonresponders if they experienced any of the following: initiated new DMARDs, biologics, or systemic immunosuppressives; increased SSZ, MTX, or HCQ above baseline dose; initiated treatment with oral, IV, or IM corticosteroids; increased the dose of oral corticosteroids above baseline dose; or discontinued treatment due to unsatisfactory therapeutic effect. Treatment failure rules were applied only through Week 16. LOCF rules were applied for partially missing data.
||At Week 16, all remaining patients in the placebo group began receiving SIMPONI ARIA® in a blinded manner.
Study design: GO-ALIVE was a global, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of SIMPONI ARIA® compared with placebo in 208 adult patients with active AS with an inadequate response or intolerance to NSAIDs. Patients had a diagnosis of definite AS for at least 3 months according to modified New York criteria. Patients had symptoms of active disease [Bath AS Disease Activity Index (BASDAI) ≥4, VAS for total back pain of ≥4, on scales of 0 to 10 cm (0 to 100 mm), and a hsCRP level of ≥0.3 mg/dL (3 mg/L)]. At Week 0, patients were randomized in a 1:1 ratio to 1 of 2 treatment groups. Subjects in the placebo group (n=103) were randomized to receive IV placebo infusions at Weeks 0, 4, and 12. At Week 16, these patients were crossed over to SIMPONI ARIA® and received administrations at Weeks 16, 20, and q8w thereafter through Week 52. Patients in the SIMPONI ARIA® group (n=105) were randomized to receive SIMPONI ARIA® 2 mg/kg infusions at Weeks 0, 4, and 12. These patients received a placebo infusion at Week 16 to maintain the treatment blind and continued to receive SIMPONI ARIA® infusions at Week 20 and q8w thereafter through Week 52. Patients were allowed to continue stable doses of concomitant MTX, SSZ, hydroxychloroquine (HCQ), low dose oral corticosteroids (equivalent 25 to ≤10 mg of prednisone per day), and/or NSAIDs during the trial. The primary endpoint was the percentage of patients achieving ASAS20 response at Week 16.